Optimization & visualization of the electron density¶
import basic stuff¶
[1]:
import numpy as np
[2]:
import sys
import os
import local dftpy module (optional)¶
[3]:
# sys.path.append('/Users/michele/Box/devel/dftpy_2/src/')
# data = '/Users/michele/Box/devel/dftpy_2/examples/DATA/'
data = '/home/sxc/work/dftpy/dftpy/examples/DATA/'
[4]:
from dftpy.field import *
from dftpy.grid import *
from dftpy.functional import LocalPseudo, Functional, TotalFunctional
from dftpy.formats import io
from dftpy.math_utils import bestFFTsize
from dftpy.time_data import TimeData
from dftpy.optimization import Optimization
from dftpy.constants import LEN_CONV, ENERGY_CONV
from dftpy.system import System
simple functions to make a grid and initial guess¶
[5]:
def MakeGrid(metric,gap):
nr = np.zeros(3, dtype = 'int32')
for i in range(3):
nr[i] = int(np.sqrt(metric[i, i])/gap)
print('The initial grid size is ', nr)
for i in range(3):
nr[i] = bestFFTsize(nr[i])
print('The final grid size is ', nr)
grid = DirectGrid(lattice=lattice, nr=nr, units=None, full=False)
return grid
def guess_rho(grid,ions):
charge_total = 0.0
zerosA = np.zeros(grid.nnr, dtype=float)
rho_ini = DirectField(grid=grid, griddata_F=zerosA, rank=1)
for i in range(ions.nat) :
charge_total += ions.Zval[ions.labels[i]]
rho_ini[:] = charge_total/ions.pos.cell.volume
return rho_ini
read input, set pseudo files, and make a grid¶
[6]:
path_pp=data
path_pos=data
file1='Al_lda.oe01.recpot'
posfile='fcc.vasp'
ions = io.read(path_pos+posfile, names=['Al'])
lattice = ions.pos.cell.lattice
metric = np.dot(lattice.T, lattice)
gap = 0.4
grid = MakeGrid(metric,gap)
The initial grid size is [19 19 19]
The final grid size is [20 20 20]
read pseudo file, generate local pseudo, and generate guess density¶
[7]:
PP_list = {'Al': path_pp+file1}
PSEUDO = LocalPseudo(grid = grid, ions=ions,PP_list=PP_list,PME=True)
rho_ini = guess_rho(grid,ions)
setting key: Al -> /home/sxc/work/dftpy/dftpy/examples/DATA/Al_lda.oe01.recpot
instance KEDF, XC and HARTREE functionals¶
[8]:
optional_kwargs = {}
KE = Functional(type='KEDF',name='x_TF_y_vW',optional_kwargs=optional_kwargs)
XC = Functional(type='XC',name='LDA')
HARTREE = Functional(type='HARTREE')
instance DFTpy energy evaluator¶
[9]:
E_v_Evaluator = TotalFunctional(KineticEnergyFunctional=KE,
XCFunctional=XC,
HARTREE=HARTREE,
PSEUDO=PSEUDO)
instance and execute DFTpy density optimizer¶
[10]:
optimization_options = {\
'econv' : 1e-6, # Energy Convergence (a.u./atom)
'maxfun' : 50, # For TN method, it's the max steps for searching direction
'maxiter' : 100,# The max steps for optimization
}
optimization_options["econv"] *= ions.nat
opt = Optimization(EnergyEvaluator=E_v_Evaluator, optimization_options = optimization_options,
optimization_method = 'TN')
new_rho = opt.optimize_rho(guess_rho=rho_ini)
Step Energy(a.u.) dE dP Nd Nls Time(s)
0 2.692153778823E+00 2.692154E+00 7.877090E-01 1 1 7.884741E-03
1 2.509905424425E+00 -1.822484E-01 7.033303E-02 2 1 1.937842E-02
2 2.502273306166E+00 -7.632118E-03 4.803902E-03 7 1 4.957438E-02
3 2.502030390983E+00 -2.429152E-04 3.640300E-04 5 1 6.849957E-02
4 2.501995476771E+00 -3.491421E-05 3.590430E-05 6 1 9.180236E-02
5 2.501992830259E+00 -2.646512E-06 2.500142E-06 5 1 1.138127E-01
6 2.501992459789E+00 -3.704704E-07 4.565581E-08 8 1 1.537414E-01
#### Density Optimization Converged ####
Chemical potential (a.u.): 0.3011519852521759
Chemical potential (eV) : 8.194762140773587
evaluate final energy, make a mol system class, and print results¶
[11]:
Enew = E_v_Evaluator.Energy(rho=new_rho, ions=ions, usePME = True)
mol = System(ions,cell=grid,name='test',field=new_rho)
print('Energy New (a.u.)', Enew)
print('Energy New (eV)', Enew * ENERGY_CONV['Hartree']['eV'])
print('Energy New (eV/atom)', Enew * ENERGY_CONV['Hartree']['eV']/ions.nat)
print('-' * 31, 'Time information', '-' * 31)
print("{:28s}{:24s}{:20s}".format('Label', 'Cost(s)', 'Number'))
for key in TimeData.cost :
print("{:28s}{:<24.4f}{:<20d}".format(key, TimeData.cost[key], TimeData.number[key]))
print('-' * 80)
Energy New (a.u.) -8.281138759120598
Energy New (eV) -225.34124199416655
Energy New (eV/atom) -56.33531049854164
------------------------------- Time information -------------------------------
Label Cost(s) Number
Optimize 0.1552 1
TF 0.0128 41
vW 0.0357 41
FFT 0.0124 84
InvFFT 0.0146 83
LDA 0.0433 41
Hartree_Func 0.0215 41
Vion_PME 0.0024 1
Ewald_Energy 0.0089 1
Ewald_Energy_corr 0.0001 1
Ewald_Energy_Real 0.0065 1
Ewald_Energy_Rec_PME 0.0023 1
_calc_PME_Qarray 0.0013 1
--------------------------------------------------------------------------------
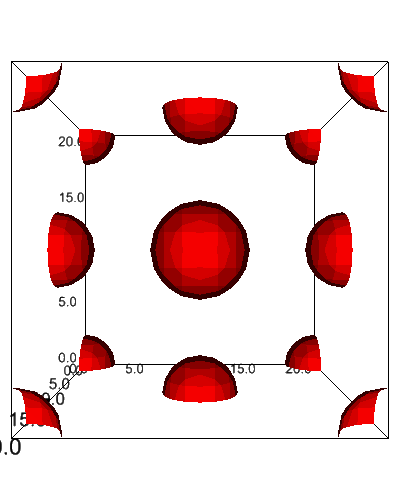
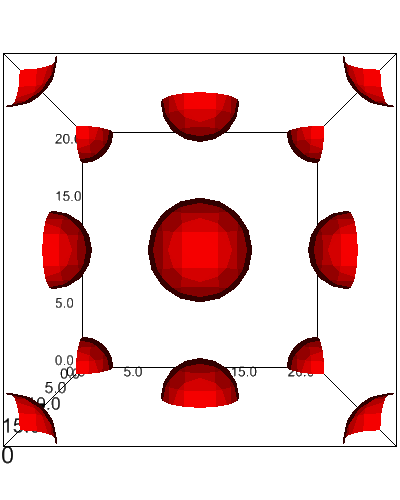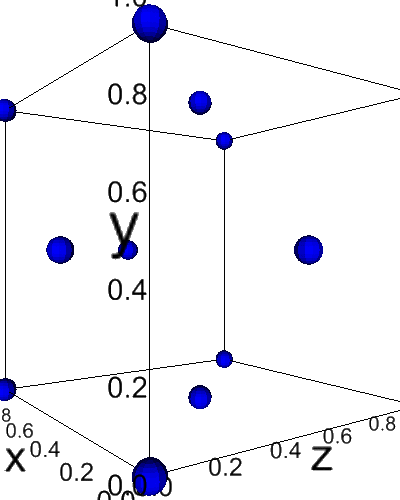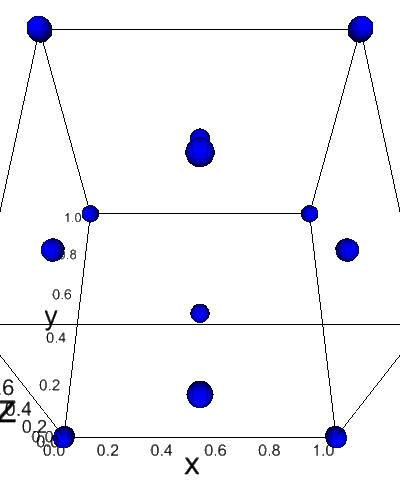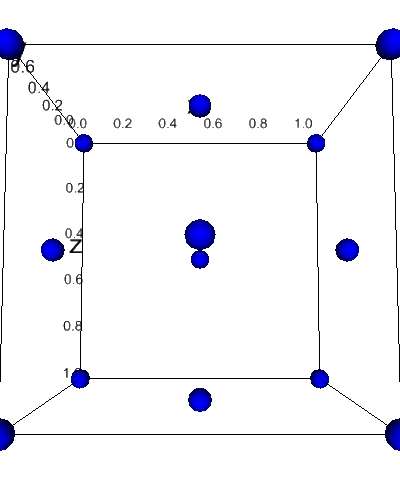
Visualize with ipv¶
[12]:
from dftpy.visualize.jupyter import view_density
view_density(mol)
/home/sxc/soft/local/venv/pure1/lib/python3.8/site-packages/ipyvolume/pylab.py:593: FutureWarning: marching_cubes_lewiner is deprecated in favor of marching_cubes. marching_cubes_lewiner will be removed in version 0.19
values = measure.marching_cubes_lewiner(data, level)
[15]:
import ipyvolume as ipv
def set_view(figure, framenr, fraction):
if fraction>0.5:
azimuth=0.0
elevation=((fraction-0.5)*2-0.5)*180
else:
azimuth=fraction*720
elevation=0.0
ipv.view(azimuth, elevation, distance=2)
ipv.movie('movie.gif', set_view, fps=20, frames=80)
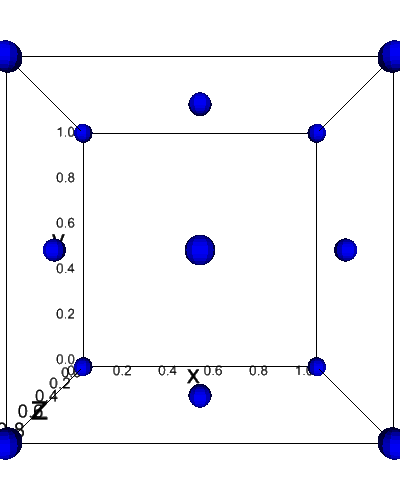
[13]:
from dftpy.visualize.jupyter import view_ions
view_ions(mol)
Visualize with VESTA¶
[14]:
from dftpy.visualize.vesta import view_on_vesta
view_on_vesta(mol)
OS is: linux
[ ]: