Molecular Dynamics Simulation¶
DFTpy performs molecular dynamics (MD) simulations with ASE_. This is one example to run NVT (canonical ensemble) simulation:
The output md.traj can be converted with ASE_:
Run this cell to install DFTpy in google colab
!python -m pip install "git+https://github.com/Quantum-MultiScale/DFTpy.git@dev"
Download the pseudopotential file
!wget https://raw.githubusercontent.com/Quantum-MultiScale/DFTpy/dev/DATA/Al_lda.oe01.recpot
[1]:
import os
import pathlib
import numpy as np
from ase.lattice.cubic import FaceCenteredCubic
from ase.md.langevin import Langevin
from ase.md.verlet import VelocityVerlet
from ase.md.velocitydistribution import MaxwellBoltzmannDistribution
from ase.io.trajectory import Trajectory
from ase import units
from ase.md.npt import NPT
from dftpy.config import DefaultOption, OptionFormat
from dftpy.interface import OptimizeDensityConf
from dftpy.api.api4ase import DFTpyCalculator
For parallel implementation when running in python
[2]:
# MPI / parallel setup
from dftpy.mpi import MP, sprint
mp = MP(parallel=False) # Set parallel=True to run in parallel
Define initial configuration with DFTpy
[3]:
conf = DefaultOption()
conf['PATH']['pppath'] = '../DATA'
conf['PP']['Al'] = 'Al_lda.oe01.recpot'
conf['OPT']['method'] = 'TN'
conf['KEDF']['kedf'] = 'WT'
conf['JOB']['calctype'] = 'Energy Force'
conf = OptionFormat(conf)
Build the system with ASE
[4]:
size = 3
a = 4.24068463425528
T = 1023 # Kelvin
T *= units.kB
atoms = FaceCenteredCubic(directions=[[1, 0, 0], [0, 1, 0], [0, 0, 1]],
latticeconstant = a,
symbol="Al",
size=(size, size, size),
pbc=True)
Set the DFTpy calculator to the ase atoms
[5]:
calc = DFTpyCalculator(config = conf) # DFTpy can be run in parallel using the MPI interface, as follows: DFTpyCalculator(config = conf, mp=mp)
atoms.calc = calc
Perform the dynamics
[ ]:
MaxwellBoltzmannDistribution(atoms, T, force_temp = True)
dyn = Langevin(atoms, 2 * units.fs, T, 0.1)
step = 0
interval = 1
def printenergy(a=atoms):
global step, interval
epot = a.get_potential_energy() / len(a)
ekin = a.get_kinetic_energy() / len(a)
print('Step={:<8d} Epot={:.5f} Ekin={:.5f} T={:.3f} Etot={:.5f}'.format(step, epot, ekin, ekin / (1.5 * units.kB), epot + ekin))
step += interval
dyn.attach(printenergy, interval=1)
traj = Trajectory('md.traj', 'w', atoms)
dyn.attach(traj.write, interval=5)
dyn.run(500)
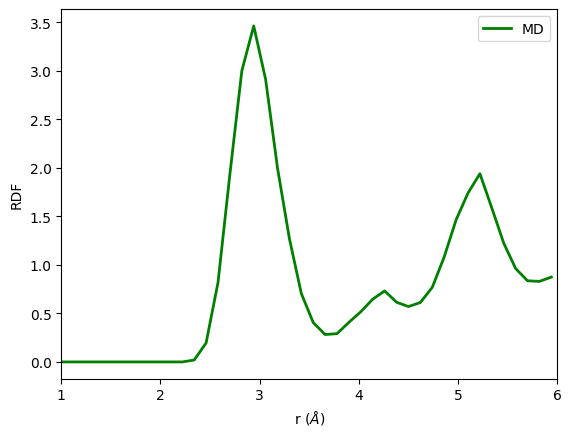
Analyse the trajectory by plotting the rdial distribution function (RDF)
[25]:
from ase.geometry.analysis import Analysis
analysis = Analysis(list(Trajectory('md.traj')))
rdf = analysis.get_rdf(rmax=6, nbins=50, return_dists=True)
rdf = np.asarray(rdf).mean(axis=0)
[20]:
import matplotlib.pyplot as plt
[28]:
plt.plot(rdf[1], rdf[0], 'g', lw=2,label='MD')
plt.ylabel('RDF')
plt.xlabel(r'r ($\AA$)')
plt.xlim(1,6)
plt.legend()
[28]:
<matplotlib.legend.Legend at 0x122f91f30>